Wilson's disease
Wilson's disease is an autosomal recessive disorder characterised by excessive copper deposition in the tissues. Metabolic abnormalities include increased copper absorption from the small intestine and decreased hepatic copper excretion. Wilson's disease is caused by a defect in the ATP7B gene located on chromosome 13.
The onset of symptoms is usually between 10 - 25 years. Children usually present with liver disease whereas the first sign of disease in young adults is often neurological disease
Features result from excessive copper deposition in the tissues, especially the brain, liver and cornea:
Diagnosis
Management
The onset of symptoms is usually between 10 - 25 years. Children usually present with liver disease whereas the first sign of disease in young adults is often neurological disease
Features result from excessive copper deposition in the tissues, especially the brain, liver and cornea:
- liver: hepatitis, cirrhosis
- neurological: basal ganglia degeneration, speech and behavioural problems are often the first manifestations. Also: asterixis, chorea, dementia
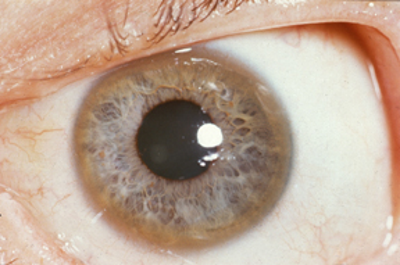
- Kayser-Fleischer rings (a brown ring on the edge of the cornea)
 | |
- renal tubular acidosis (esp. Fanconi syndrome)
- haemolysis
- blue nails
Diagnosis
- reduced serum caeruloplasmin
- reduced serum copper (counter-intuitive, but 95% of plasma copper is carried by ceruloplasmin)
- increased 24hr urinary copper excretion
Management
- penicillamine (chelates copper) has been the traditional first-line treatment
- trientine hydrochloride is an alternative chelating agent which may become first-line treatment in the future
- tetrathiomolybdate is a newer agent that is currently under investigation

